首页 有限元仿真 结构强度分析 流体CFD仿真 热仿真分析 电磁场分析 离散元仿真 声学光学化学 分子动力学 分子对接计算 分子互作模拟 药物相关模拟 动力学后数据 第一性原理 电子性质计算 结构性质计算 材料性质计算 催化性质计算 能量性质计算 光声磁性质计算 量子化学 分子性质预测 化学反应机理 光谱预测计算 弱相互作用 激发态反应 行业案例 新闻动态 公司新闻 行业动态 技术学堂 关于我们 公司简介 人才招聘 联系我们 186 6240 7040
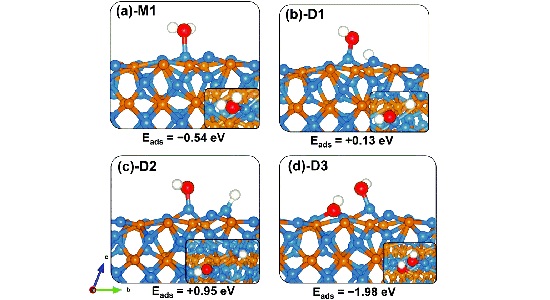
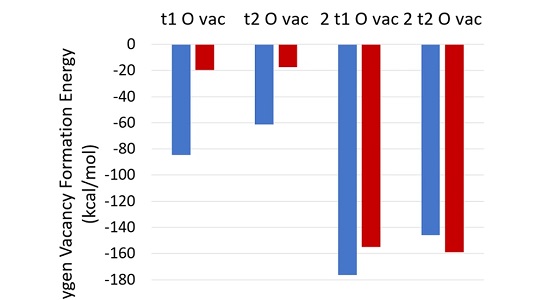
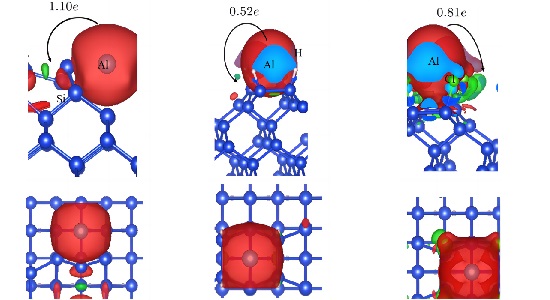
TAM-TFTA-COF吸附分子机理分析 TAM-TFTA-COF吸附分子机理分析本工作拟基于CP2K软件进行计算,第一性原理是指不使用经验参数,只通过基本物理常量(me、c、e、h、kB)来计算微观体 结合能计算,形成能计算 结合能计算结合能(Binding energy)是指在核物理中,原子核内的核子通过核力相互结合形成原子核时释放或吸收的能量。它表示了将离散的核子聚集成稳定原子核 Bader电荷计算 Bader电荷计算计算Bader电荷来得到原子周围的电子数,从而近似得到原子的化合价。Bader电荷分析是理査德·贝德(RichardBader)开发的种将分子 吉布斯自由能 吉布斯自由能吉布斯自由能可以用于判断化学反应和相变是否会发生。当系统发生化学反应或相变时,吉布斯自由能会发生变化。 吉布斯自由能是热力学中的重要概念,用于预测和 差分电荷密度 差分电荷密度养分电荷容度(Dierenial Chare Densiy)是指在化学反应或分子间相互作用中,电子容度的变化。它描述了电子在空间上的重新分布,反映了 迁移能垒,吸附能 迁移能垒迁移能垒计算是通过计算机模拟来研究材料中原子迁移的过程。这种计算通常使用分子动力学模拟或者密度泛函理论方法。在议种计算中,先要给定材料的晶体结构以及化学 共1页 6条
186 6240 7040 2746 12474@qq.com 江苏省苏州市吴中区胥口镇顾巷路228号 关注官方公众号 有限元仿真 结构强度分析 流体CFD仿真 热仿真分析 电磁场分析 离散元仿真 声学光学化学 分子动力学 分子对接计算 分子互作模拟 药物相关模拟 动力学后数据 第一性原理 电子性质计算 结构性质计算 材料性质计算 催化性质计算 能量性质计算 光声磁性质计算 量子化学 分子性质预测 化学反应机理 光谱预测计算 弱相互作用 激发态反应



 关注官方公众号
关注官方公众号